医疗器械产品取得注册证和生产许可证后,就可以上市销售。企业产品上市后,需要按照法规的要求进行生产和销售活动。现阶段医疗器械的监管包括上市前审批和上市后监管,从监督角度来说,上市后的监管比准入审批更重要。依据《医疗器械监督管理条例》,如上市后发现企业有不规范经营,则可能接受相应的处罚。
医疗器械上市后,为保证产品质量,企业需重点关注顾客投诉、不良事件、质量管理体系运行情况、法规标准变化,以及产品抽检和飞行检查。以下对几个重要的事项做出说明。
1、医疗器械不良事件监测
国家医疗器械不良事件监测信息系统注册要求:注册人、经营企业和二级以上医疗机构应当注册为国家医疗器械不良事件监测信息系统用户,主动维护其用户信息,报告医疗器械不良事件。注册人应当持续跟踪和处理监测信息;产品注册信息发生变化的,应当在系统中立即更新。
医疗器械注册人在取得医疗器械注册证或备案凭证后,需要在“国家医疗器械不良事件监测信息系统”申请账号、设置密码。
网址为:https://maers.adrs.org.cn/console/login.ftl,登录界面如下:
新开办的企业点击“注册”,跳转到注册界面,按实际情况填写并提交,生产企业用户类型为“持有人”,如下:

账号注册完成以后,即可登录网址,登录后需要完善已注册或备案的产品信息,界面如下:
图|不良事件监测信息系统查询界面
企业可通过对应的栏目,选择上报不良事件,或查询相关单位上报的不良事件信息。
医疗器械注册人需要指定专人负责不良事件监测工作,定期登录网站查询是否有不良事件发生。如有,需及时和申报人进行沟通,了解不合格情况,并对问题进行处理,必要时执行纠正和预防措施。
2、年度医疗器械质量管理体系自查报告
《医疗器械监督管理条例》规定,医疗器械注册人每年需要向药监部门提交年度医疗器械质量管理体系自查报告,报告的填报内容为当年度1月1日至12月31日统计数据,应于次年3月31日之前提交。
如当地药监部门没有特别说明,自查报告均在国家药监局官网上申报提交,注册人需要在国家药监局官网上注册账号,见下图:
图|国家药品监督管理局注册登录界面
注册账号后,登录网页,点击“我的绑定”→“医疗器械生产企业监管信息系统”,见下图:
打开界面后,需要完善企业基本信息和产品基本信息。申报质量管理体系自查报告时,点击“年度自查报告”→“新增”,按界面要求填报,见下图:

3、医疗器械内审管理评审
按照质量管理体系《程序文件》的要求,企业每12个月至少进行一次内部审核和管理评审,如有必要,可增加内审和管理评审的次数。
内部审核由管理者代表组织,内审员负责对各部门进行审核,审核内容需要包含《医疗器械生产质量管理规范现场指导原则》或相应附录的内容。内审员需要对发现的不合格项的纠正和预防进行跟进,直至不合格项关闭。如有必要,内部审核可引入第三方进行审核。
管理评审由总经理组织,对本年度的质量管理体系运行情况进行总结和改进,需要形成管理评审记录。
4、医疗器械法规标准更新
医疗器械的监管是基于相应的法律法规和标准进行,如法规和标准有更新,注册人需要及时进行学习解读,保证产品性能、质量管理水平能符合要求。医疗器械上市后,需要制定专人关注法规和标准更新情况,法规和标准的发布更新可通过查询国家药监局等网站进行查询,如下图:

图|2023年国家药监局发布的医疗器械行业标准
法规一般发布即实施,或者半年到一年的缓冲期,而标准发布后一般有2-3年的缓冲期,企业需要关注和产品相关的强制性国际标准、行业标准的实施情况,保证产品在标准实施后能符合要求。以有源产品相关的GB9706相关标准为例,未来3年内,有一批强标需要更新,见下表:
表 即将实施的9706系列标准目录
| 序号 | 产品类型 | 实施日期 |
| GB 9706.204-2022 | 医用电气设备 第2-4部分:心脏除颤器的基本安全和基本性能专用要求 | 2024-08-01 |
| GB 9706.222-2022 | 医用电气设备 第2-22部分:外科、整形、治疗和诊断用激光设备的基本安全和基本性能专用要求 | 2024-05-01 |
| GB 9706.255-2022 | 医用电气设备 第2-55部分:呼吸气体监护仪的基本安全和基本性能专用要求 | 2026-01-01 |
| GB 9706.271-2022 | 医用电气设备 第2-71部分:功能性近红外光谱(NIRS)设备的基本安全和基本性能专用要求 | 2026-01-01 |
| GB 9706.275-2022 | 医用电气设备 第2-75部分:光动力治疗和光动力诊断设备的基本安全和基本性能专用要求 | 2026-01-01 |
| GB 9706.283-2022 | 医用电气设备 第2-83部分:家用光治疗设备的基本安全和基本性能专用要求 | 2026-01-01 |
| GB 9706.290-2022 | 医用电气设备 第2-90部分:高流量呼吸治疗设备的基本安全和基本性能专用要求 | 2026-01-01 |
| YY 9706.221-2021 | 医用电气设备 第2-21部分:婴儿辐射保暖台的基本安全和基本性能专用要求 | 2024-05-01 |
| YY 9706.230-2023 | 医用电气设备 第2-30部分:自动无创血压计的基本安全和基本性能专用要求 | 2026-01-15 |
| YY 9706.231-2023 | 医用电气设备 第2-31部分:带内部电源的体外心脏起搏器的基本安全和基本性能专用要求 | 2026-05-01 |
| YY 9706.234-2021 | 医用电气设备 第2-34部分:有创血压监护设备的基本安全和基本性能专用要求 | 2024-05-01 |
| YY 9706.246-2023 | 医用电气设备 第2-46部分: 手术台的基本安全和基本性能专用要求 | 2026-01-15 |
| YY 9706.247-2021 | 医用电气设备 第2-47部分:动态心电图系统的基本安全和基本性能专用要求 | 2024-05-01 |
| YY 9706.249-2023 | 医用电气设备 第2-49部分:多参数患者监护仪的基本安全和基本性能专用要求 | 2026-01-15 |
| YY 9706.252-2021 | 医用电气设备 第2-52部分:医用病床的基本安全和基本性能专用要求 | 2024-05-01 |
| YY 9706.256-2023 | 医用电气设备 第2-56部分:用于体温测量的临床体温计的基本安全和基本性能专用要求 | 2026-05-01 |
| YY 9706.258-2022 | 医用电气设备 第2-58部分:眼科手术用晶状体摘除及玻璃体切除设备的基本安全和基本性能专用要求 | 2025-06-01 |
| YY 9706.261-2023 | 医用电气设备 第2-61部分:脉搏血氧设备的基本安全和基本性能专用要求 | 2026-01-15 |
| YY 9706.264-2022 | 医用电气设备 第2-64部分:轻离子束医用电气设备的基本安全和基本性能专用要求 | 2025-06-01 |
| YY 9706.268-2022 | 医用电气设备 第2-68部分:电子加速器、轻离子束治疗设备和放射性核素射束治疗设备用的X射线图像引导放射治疗设备的基本安全和基本性能专用要求 | 2025-06-01 |
| YY 9706.270-2021 | 医用电气设备 第2-70部分:睡眠呼吸暂停治疗设备的基本安全和基本性能专用要求 | 2024-05-01 |
| YY 9706.272-2021 | 医用电气设备 第2-72部分:依赖呼吸机患者使用的家用呼吸机的基本安全和基本性能专用要求 | 2024-05-01 |
| YY 9706.274-2022 | 医用电气设备 第2-74部分:呼吸湿化设备的基本安全和基本性能专用要求 | 2025-05-01 |
| YY 9706.277-2023 | 医用电气设备 第2-77部分:采用机器人技术的辅助手术设备的基本安全和基本性能专用要求 | 2026-01-15 |
|
YY 9706.278-2023 |
医用电气设备 第2-78部分:康复、评定、代偿或缓解用医用机器人的基本安全和基本性能专用要求 | 2026-05-01 |
| YY 9706.279-2023 | 医用电气设备 第2-79部分:用于呼吸功能障碍的呼吸支持设备的基本安全和基本性能专用要求 | 2026-05-01 |
| YY 9706.280-2023 | 医用电气设备 第2-80部分:用于呼吸功能不全的呼吸支持设备的基本安全和基本性能专用要求 | 2026-05-01 |
| YY 9706.284-2023 | 医用电气设备 第2-84部分:紧急医疗服务环境用呼吸机的基本安全和基本性能专用要求 | 2026-05-01 |
5、医疗器械飞行检查
国家局发布的《关于加强医疗器械生产经营分级监管工作的指导意见》提出,对医疗器械生产企业按等级进行监管,监督的方式以飞行检查为主,飞行检查即不通知的突击检查。见下表:
| 监管级别 | 监管对象 | 监管频次 |
| 四级监管 | 本行政区域重点监管品种目录产品,以及质量管理体系运行状况差、有严重不良监管信用记录的企业。 | 每1年全项目检查≥1次 |
| 三级监管 | 除本行政区域重点监管品种目录以外第三类医疗器械,以及质量管理体系运行状况较差、有不良监管信用记录的企业。 | 每1年检查≥1次
每2年全项目检查≥1次 |
| 二级监管 | 除本行政区域重点监管品种目录以外第二类医疗器械的企业。 | 每2年检查≥1次 |
| 一级监管 | 第一类医疗器械生产企业。 | 每年随机抽取本行政区域25%以上的企业 |
全项目检查是指药品监督管理部门按照医疗器械生产质量管理规范及相应附录,对监管对象开展的覆盖全部适用项目的检查。对委托生产的医疗器械注册人备案人开展的全项目检查,应当包括对受托生产企业相应生产活动的检查。
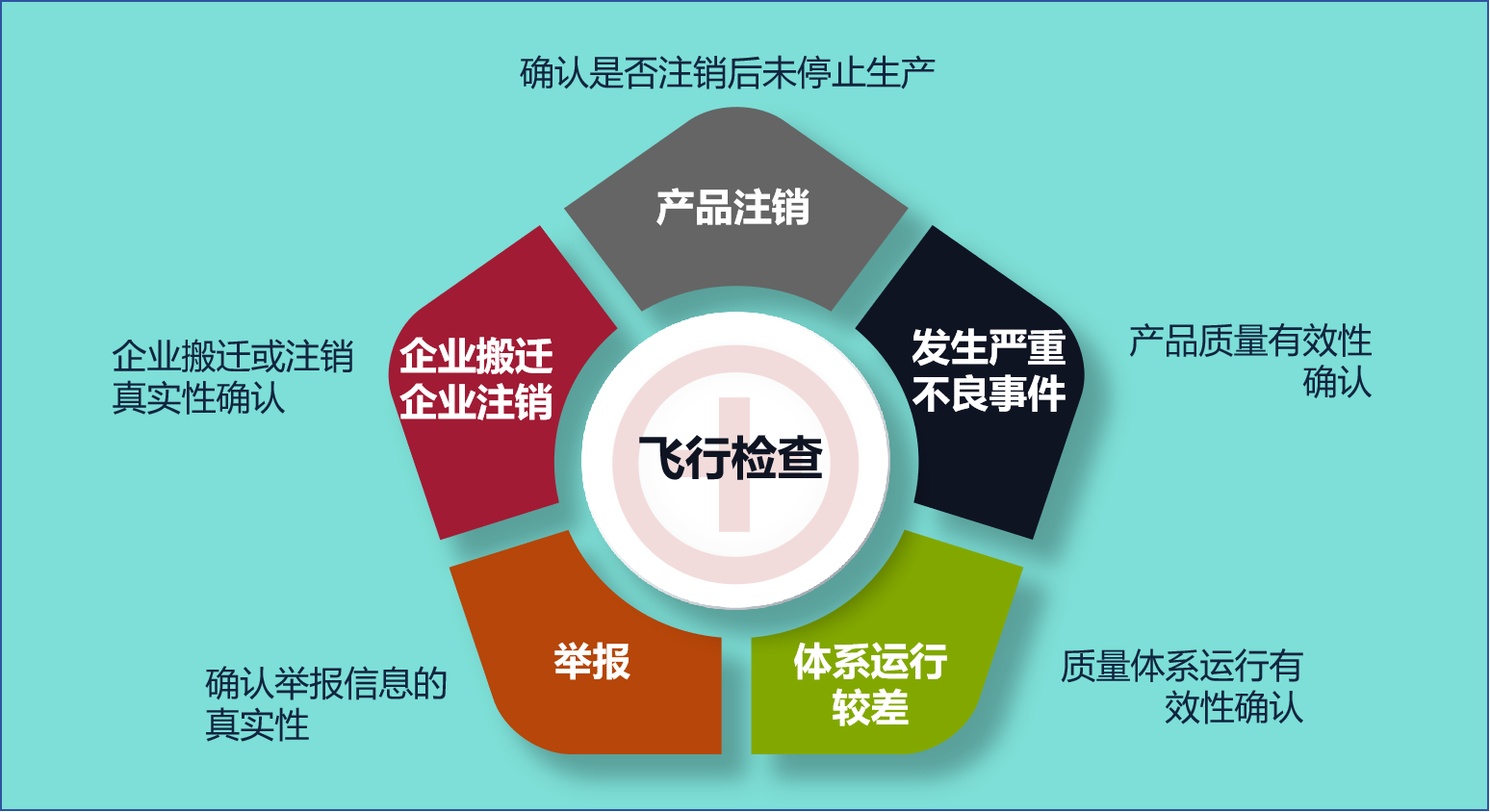
企业需要在取得注册证后,依旧严格保证已建立的质量管理体系正常运行,确保文件和记录完整有效。某些情况下,企业更容易被飞行检查,见下图: