
《医疗器械生产质量管理规范》中规定“应当根据产品和工艺特点制定留样管理规定,按规定进行留样,并保持留样观察记录”。实际操作过程中,企业如何界定产品的具体留样管理方式?本结合体系核查的实践,对医疗器械产品留样管理进行说明,为医疗器械生产企业开展产品留样管理提供参考。
医疗器械产品留样定义指南

产品留样指生产企业按照规定保存的、用于质量追溯或调查以及产品性能研究的物料、产品样品。产品留样在医疗器械产品质量追溯、不良事件调查中有助于查找问题、明晰事故责任,也可为确认或修改产品技术指标提供数据支持。
医疗器械生产企业应当根据产品特性、工艺特点、临床应用等,明确产品留样的目的。留样目的不同,留样方式也将不同,包括每批留样和单次留样,医疗器械GMP审查主要关注每批留样的情况:
- 每批留样
用于医疗器械产品质量追溯,保证每批次产品可追溯项目,如无菌性能、理化性能。每批产品(或关键原材料)均需留样。 - 单次留样
用于稳定性研究,生产企业开发新产品、新工艺或变更产品有效期等指标时,用于考察产品稳定性。只需要研发阶段留一次样品。
医疗器械留样管理要求
企业应当根据产品特点和工艺特点制定留样管理制度,明确留样目的、留样样品、留样比例或数量、留样观察等方面的要求,开展留样观察,并保持相关记录。要点如下:
1)留样室:每批留样的产品需要有独立的留样室,留样室环境控制要求同成品仓库。单次留样的产品可以使用留样柜进行存放。
2)留样样品:最终成品。
3)留样数量:至少满足一次全性能检验的数量。
4)留样观察:定期检查产品外观,失效前进行一次全性能检验,环氧乙烷残留量可以不检验。
留样时间:不少于每批产品的有效期。
哪些医疗器械需要每批留样?
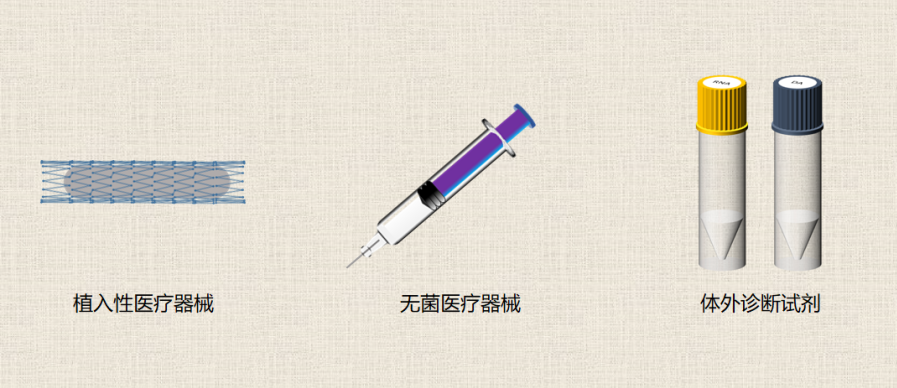
医疗器械使用时会改变产品性质的产品需要留样,以便于追溯产品的出厂状态,因此以下三类产品需要留样:
1)无菌医疗器械(或有微生物限度要求的产品):使用后会改变产品的无菌状态。
2)植入性医疗器械:使用后会改变产品的无菌状态、产品植入后改变性质。
3)体外诊断试剂:使用后产品性状改变。
哪些医疗器械不需要每批留样?

医疗器械使用时不会改变产品性质的产品可以不留样,追溯产品时可直接追溯到对应的产品,因此以下三类产品可不留样:
- 设备类医疗器械:产品可重复使用,且使用前后不会改变产品的技术指标。
- 非无菌类医疗器械(包括医疗器械软件):使用前后不会改变产品的技术指标。
- 定制式医疗器械:可重复使用,使用前后不会改变产品的技术指标。