
医疗器械企业在生产销售第二类医疗器械时,需要取得第二类医疗器械注册证。企业在注册时,如何判断产品是不是第二类医疗器械,第二类医疗器械注册流程是什么,第二类医疗器械注册需要什么条件,本文将解答以上几个关键问题。
1、怎么区分第二类医疗器械?
医疗器械是指用于人体治疗或诊断的仪器、设备、器具、体外诊断试剂及校准物、材料以及其他类似或者相关的物品,包括所需要的计算机软件。
医疗器械依据风险等级,分为第一类、第二类、第三类医疗器械,第二类医疗器械为中风险的医疗器械。
可通过查询《医疗器械分类目录》来明确具体的产品分类,如果分类目录中无该产品,可通过向省级药监部门提交分类界定,由药监局确定具体的分类。

一般接触皮肤、创伤组织,使用时可能有轻微损伤、中度损伤的器械,有较大可能性为第二类医疗器械。
2、第二类医疗器械怎么注册?
国内生产的第二类医疗器械产品,需要向所在地省级药监局申请《医疗器械注册证》。并向市级市场监督管理局申请《医疗器械生产许可证》。
医疗器械注册过程通过产品分析、技术要求编写、注册检验、临床评价、注册资料编写申报、建立质量管理体系等过程,通过注册审批,取得《医疗器械注册证》。第二类医疗器械注册过程见下图:
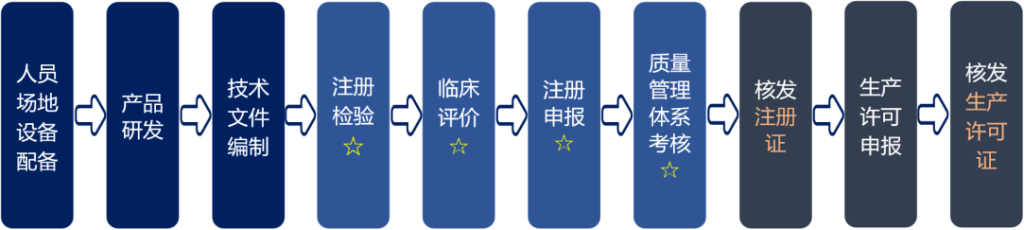
- 注册检验
第二类医疗器械样品生产完之后,需要将产品送至各省医疗器械检验所或有资质的第三方检测机构,按照《产品技术要求》进行注册检验,并获得检验报告。 - 临床评价
如第二类医疗器械产品在《免于临床评价的医疗器械目录》中,则可以不进行临床试验。
如不在目录中,但是能获得已取证的同类产品技术要求和临床文献,则可以通过同品种比对完成临床评价。
如无法取得相应材料,则需要进行临床试验。 - 注册申报
按照注册申报资料格式要求,编写相应的医疗器械注册申报材料,国产第二类医疗器械注册提交到对应省级药品监督管理局,进口第二类医疗器械注册提交到国家药监局进行注册。 - 质量管理体系
第二类医疗器械生产企业需要依据YY/T 0287-2007、《医疗器械生产质量管理规范》建立和实施医疗器械质量管理体系(GMP),体系需要运行3个月以上。药监部门会在注册审评过程中组织对企业进行质量管理体系现场检查。
3、第二类医疗器械注册需要什么条件?
第二类医疗器械注册生产条件主要包括人员、场地和设施设备。
1)人员
企业需要根据部门设置,适当配备人员,第二类医疗器械生产企业至少应当配置7-10人。人员要求如下:
| 序号 | 职能部门 | 人员配置 | 任职要求 | 岗位职责 |
| 1 | 总经理 | 总经理 | / | 最高负责人 |
| 2 | 管理者代表 | 管理者代表 | 执行力强 | 医疗器械项目管理 |
| 3 | 生产部 | 生产负责人 | 大专以上学历 | 负责产生管理 |
| 4 | 操作工 | 若干人 | 干活 | |
| 5 | 质量部 | 质量负责人 | 大专以上学历 | 负责检验管理 |
| 6 | 检验员 | ≥2人 | 检验 | |
| 7 | 研发部 | 技术负责人 | 大专以上学历 | 负责产品研发 |
| 8 | 采购部 | 采购负责人 | 读过书,有文化 | 负责采购理 |
| 9 | 销售部 | 销售负责人 | 负责销售管理 | |
| 10 | 行政部 | 行政负责人 | 负责行政管理 | |
| 11 | 仓库 | 仓库管理员 | 负责仓库管理 |
2)场地
医疗器械生产企业应根据产品性质和生产规模,选择合适大小的生产场地,其中注册地址和生产地址可以在一起也可以分开。
非无菌第二类医疗器械场地面积一般要求≥300㎡,无菌第二类医疗器械场地面积一般要求≥800㎡。
3)设施设备
第二类医疗器械生产需要配备和产品相关的生产设备和检验设备,非无菌医疗器械需要配备车间、仓库、检验室等设施,无菌医疗器械需要配备洁净车间、微生物检验室、仓库等设施。
无菌医疗器械的洁净车间一般要求为十万级洁净区。
4)没有场地能不能注册第二类医疗器械
如果没有生产场地,可以走医疗器械注册人制度,找一家有类似产品生产能力的企业一起注册。需要注意以下几点:
- 科研机构、经营企业都可以申请医疗器械注册证,目前没有对个人开放。
- 需要和生产企业联合申请,生产企业需要有类似产品生产能力。
- 注册人需要有办公科研场地,配备研发、销售、质量管理人员。
- 注册人需要有专业的第三方机构指导注册以及上市后管理。